
|
Etiology/ Causes |
Williams Syndrome (WS) is a rare genetically based neurodevelopmental disorder. People who have WS are missing genetic materials from chromosome 7. The deletion is caused by a break in the DNA molecule that makes up the chromosome. The chromosome break occurs while the sperm or egg cell (the male or female gamete) is developing. When this gamete is fertilized (at conception), the child will develop Williams Syndrome. The parent, however, does not have the break in any other cells and does not have the syndrome. In fact, the break is usually such a rare event that it is very unlikely to happen again if the parent has another child.
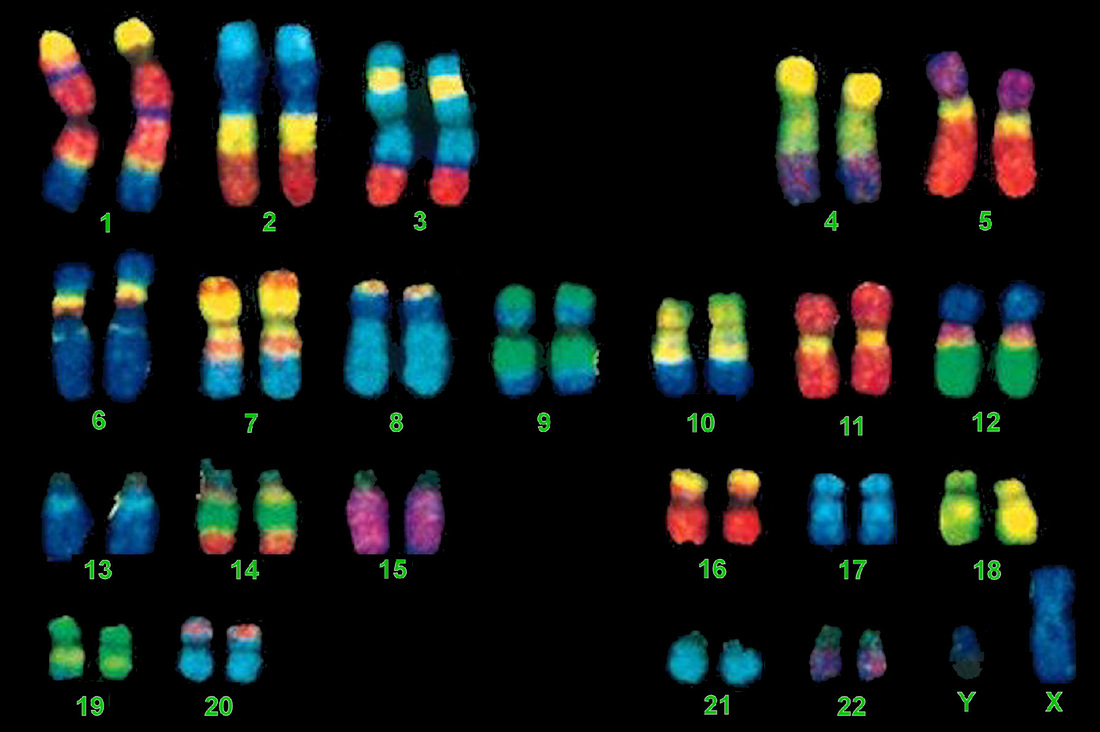
A chromosome is a single, very long strand of DNA, tightly coiled up. The largest, chromosome 1, contains about 8000 genes while the smallest, chromosome 21, contains less than 300. In all, you have 23 pairs of chromosomes in most cells of your body. You can see them under an ordinary microscope, but you would not be able to distinguish individual genes. Scientists can treat chromosomes to reveal characteristic 'banding patterns'. These patterns help them to identify each chromosome, and to spot any missing or duplicated regions.
|
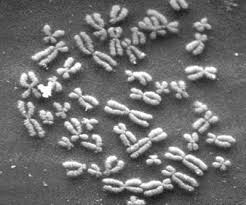
Human Chromosomes under a microscope.
Williams syndrome is considered a microdeletion syndrome, because the deletion is too small to be seen with a microscope (fewer than 5 million bases of DNA are deleted).
|
|
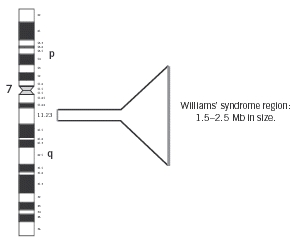
This microdeletion syndrome is caused by a chromosomal deletion spanning approximately 25-28 contiguous genes on the long (q) arm of one of the two copies of chromosome 7.
Individuals with WS may have some or all of these genes deleted. Contiguous refers to the fact that these genes are arranged next to each other. The size of the deletion can be large or small, which may explain why some individuals with WS are more severely affected than others. To the left is a schematic demonstrating the common deletion found in Williams' syndrome, at region 7q11.23, approximately 1.5-2.5 Mb (million base pairs) in size. It is thought that the missing genes in this region are important causes of the physical and mental findings of Williams Syndrome (Schubert, 2009). |

|
Gene Effects |
|
Two missing genes in particular, ELN and LIMK1, have been shown to be important in causing some of the characteristic symptoms of Williams Syndrome.
The ELN gene codes are for a protein called elastin. The job of elastin in the human body is to provide elasticity to the connective tissues such as those in the arteries, joints and tendons. This tissue is designed to stretch when an organ, like blood vessels or skin need to adjust in size. The elastin protein is made only during embryonic development and childhood, when blood vessels are formed. Researchers have found that loss of the ELN gene is associated with the connective tissue abnormalities and cardiovascular disease (specifically supravalvular aortic stenosis) found in many people with this disease (Ewart et al., 1993; Metcalfe et al., 2000). ELN is the main gene that is used to genetically diagnose Williams Syndrome. This gene is missing in 99% of individuals diagnosed with WS. |

|

|
Most individuals with Williams Syndrome are missing the LIMK1 (LIM-kinase1) gene.
This gene is associated with our ability to see an object and mentally assemble all the parts into a whole - which is called visuo-spatial cognition. The absence of this makes it difficult for someone to draw and write. Although this gene plays a contributory role in visuospatial functioning, it must be in combination with other proteins that act together to impair spatial functioning (Cornish & Wilding, 2010). The specific contribution of each of the remaining genes in the clinical picture of the disease is not yet conclusive.
List of Genes Related to Williams Syndrome. Nevertheless, CLIP2 (CYLN2), LIMK1, GTF2I, and GTF2IRD1 are strongly suspected as being the most important contributors in the cognitive and neurobehavioral phenotypes of the disease (Frangiskakis et al., 1996; Hirota et al., 2003; Morris et al., 2003; van Hagen et al., 2007; Schubert, 2009). |

|
Theories about the cause of the genetic disorder |
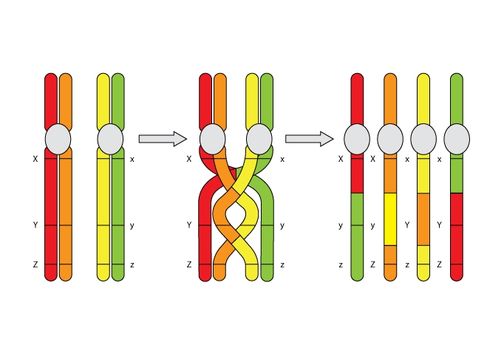
During meiosis, homologous chromosomes (1 from each parent) pair along their lengths. The chromosomes cross over at points called chiasma. At each chiasma, the chromosomes break and rejoin, trading some of their genes. This recombination results in genetic variation.
(The University of Waikato, 2008)
Deletions that happen during egg and sperm formation are caused by unequal recombination. Recombination normally occurs between pairs of chromosomes during meiosis. If the pairs of chromosomes don't line up correctly, or if the chromosome breaks aren't repaired properly, the structure of the chromosome can be altered.
Unequal recombination occurs more often than usual at this location on chromosome 7, likely due to some highly repetitive DNA sequence that flanks the commonly deleted region. Copy number variation at the 7q11.23 segmental duplications is a susceptibility factor for the Williams-Beuren syndrome deletion (Cuscó, 2008). |
Paracentric inversion of the WBS chromosomal region (7q11.23) is known as a genomic polymorphism in about 5% of the general population. This inversion is found in 25-33% of unaffected transmitting parents and appears to be a major predisposing factor for the WBS-related deletion (Osborne et al., 2001; Bayés et al., 2003; Hobart et al., 2010).
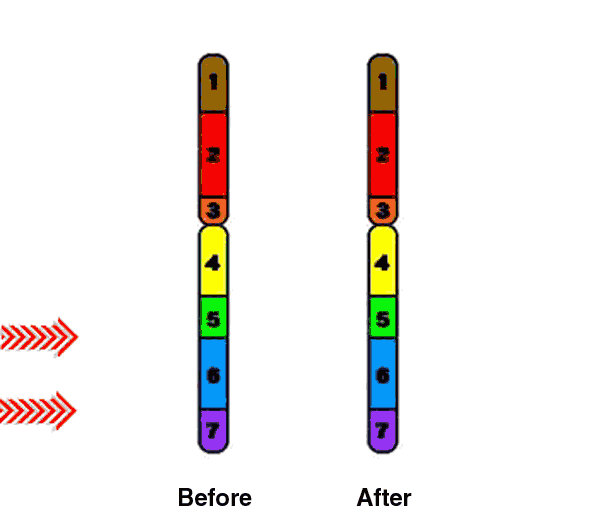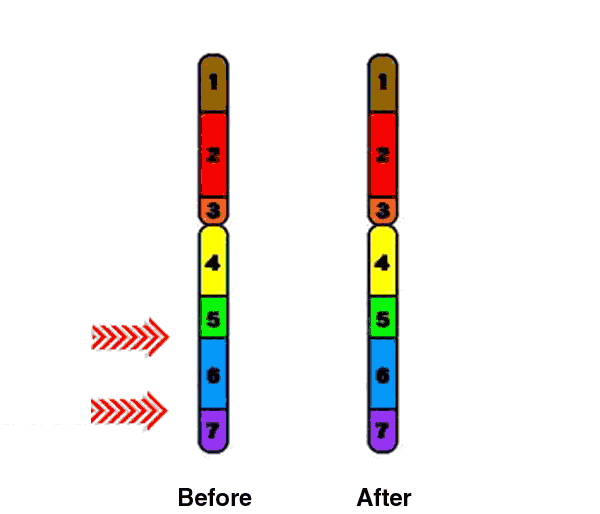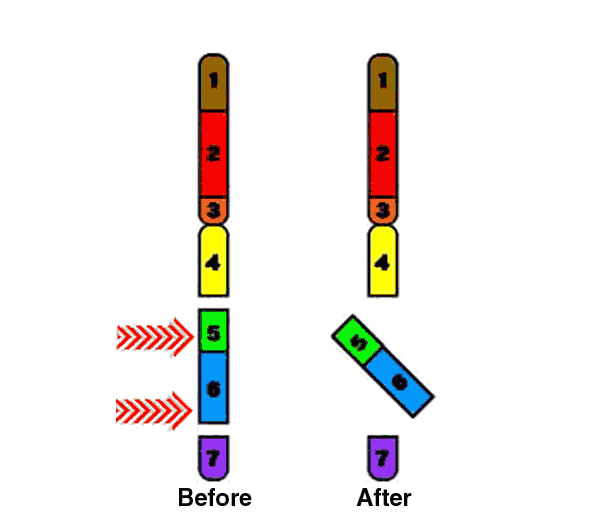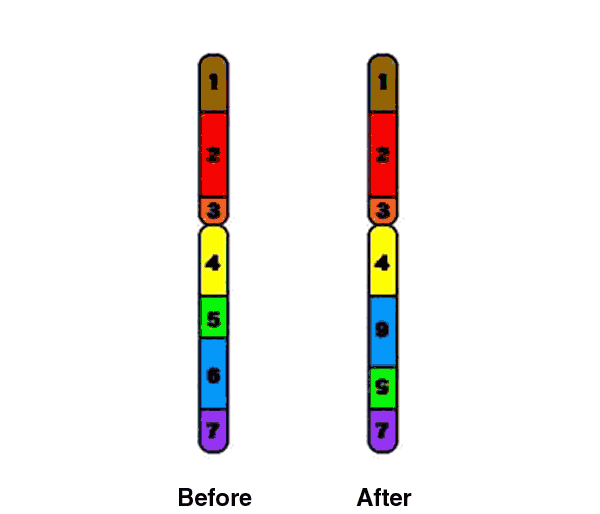
Paracentric inversion (does not include centromere)
|

|
Evaluation Process used to determine if the disorder exists. |
|
STEP 1: Clinical evaluation
|

|


|
STEP 2: Medical/Genetic Testing (i.e. a blood test)
TECHNIQUE A: It is commonly known as the FISH test* (Lowery et al. 1995) (Fluorescent In Situ Hybridization). It gets results the fastest out of all the known techniques and is relatively inexpensive. FISH is a specialized chromosome analysis. In the lab, the gene Elastin (which is only found on chromosome 7) is labelled with a fluorescent chemical (known as a probe). A control probe is also added to find chromosome 7 easier. When the genes are exposed to ultraviolet (UV) light, only the elastin gene (and control probe) will light up. An individual who doesn't have WS will have a pair of elastin genes which will light up. A person affected with Williams Syndrome will have only one elastin gene light up because the other was deleted. 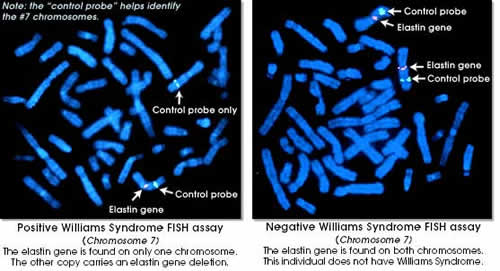
|

Research has shown that 99% of individuals with Williams Syndrome will only have one of the elastin genes show up on a FISH test.
Note 1: This FISH test is not approved by the Food and Drug Administration so it is important to confirm Williams syndrome (WS) by other established methods, such as clinical evaluation.
Note 2: Patients with a deletion outside of the elastin gene could display normal development of connective tissue, including the heart, but have other features of WS.
*The cytogenetics laboratory will need 5 ml of blood drawn in a Sodium heparin tube. The sample should arrive in the lab the same day it was drawn or on the following day. Results are usually available in 2-4 weeks.
Note 1: This FISH test is not approved by the Food and Drug Administration so it is important to confirm Williams syndrome (WS) by other established methods, such as clinical evaluation.
Note 2: Patients with a deletion outside of the elastin gene could display normal development of connective tissue, including the heart, but have other features of WS.
*The cytogenetics laboratory will need 5 ml of blood drawn in a Sodium heparin tube. The sample should arrive in the lab the same day it was drawn or on the following day. Results are usually available in 2-4 weeks.
|
Atypical individuals who have Williams Syndrome are due to a gene mutation within or near the elastin gene. These mutations would not be detected by the standard FISH test as it involves a DNA probe which only detects large deletions such as the entire elastin gene. Therefore, the FISH test may come back with normal results, completely missing the small deletions or mutations that can cause Williams Syndrome.
|
|

|
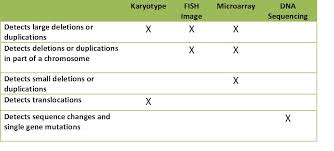
TECHNIQUE B: Chromosomal Microarray
|
Prenatal Chromosomal Microarray Analysis (Prenatal CMA) is a diagnostic test that offers pregnant women and their doctors the capability to detect fetal genetic abnormalities that had previously been impossible.
Using amniotic fluid or CVS (Chorionic villus sampling), it is possible to analyze all chromosomes in a single test.
The sensitivity and specificity of Prenatal CMA is much higher than the older technique of karyotype analysis.
Using amniotic fluid or CVS (Chorionic villus sampling), it is possible to analyze all chromosomes in a single test.
The sensitivity and specificity of Prenatal CMA is much higher than the older technique of karyotype analysis.
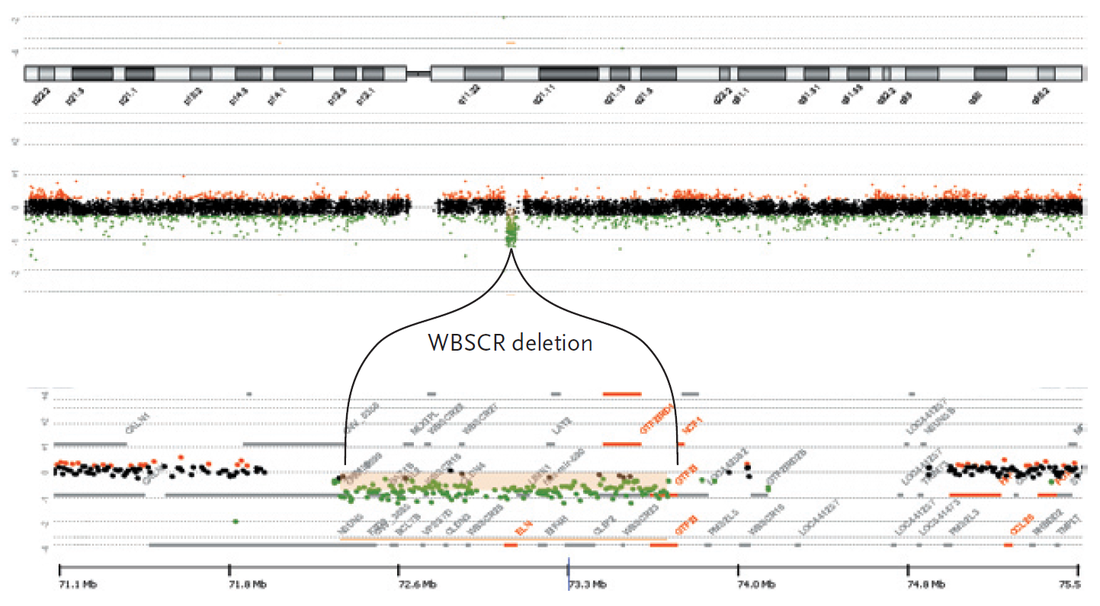
The results of array comparative genomic hybridization are shown on a schematic of chromosome 7 (top panel; from an Agilent 244K microarray), revealing the loss of one copy of the Williams-Beuren syndrome chromosome region (WBSCR), approximately 1.5Mb in size, as indicated by the cluster of green hybridization signals (middle panel). An enlarged view of the WBSCR is also shown (bottom panel).
(Pober, 2010).

|
Treatment |
|
If it is found that your child has Williams Syndrome based on an examination and genetic test, the genetics team and your primary doctor will counsel you on what that means for the child and the family. While there is no cure for Williams Syndrome it is important to find and treat the different medical problems and symptoms that can occur with this disorder.
As Williams Syndrome is a multi-system disorder, the expertise of a number of specialists is required for management of this disorder. Children with Williams Syndrome need costly and ongoing medical care and early interventions through a number of different specialists to ensure proper functioning of all parts of the body. This often includes ophthalmology, cardiology, otolaryngology, speech therapy, occupational therapy, audiology, neurology, psychology, physiotherapy and gastroenterology. |


|
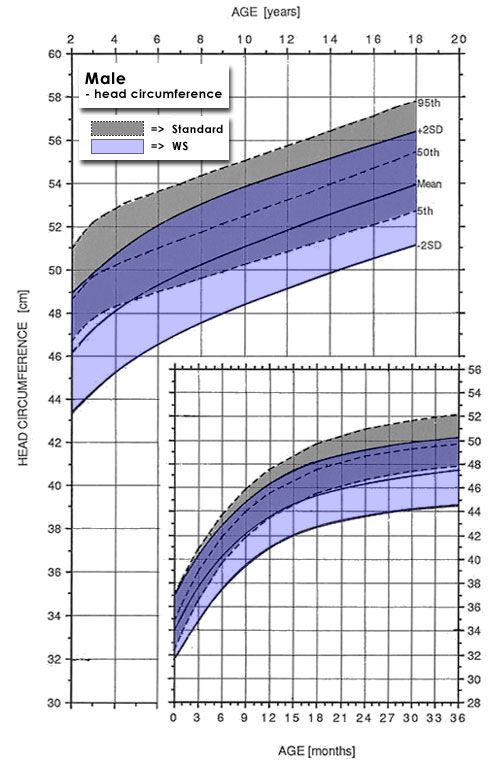
WS growth chart for males.
|
The height and growth of individuals with Williams Syndrome should be monitored using special growth curves developed specifically for individuals with Williams Syndrome. Individuals who fall off these growth curves should be assessed for possible eating or thyroid disorders.
|
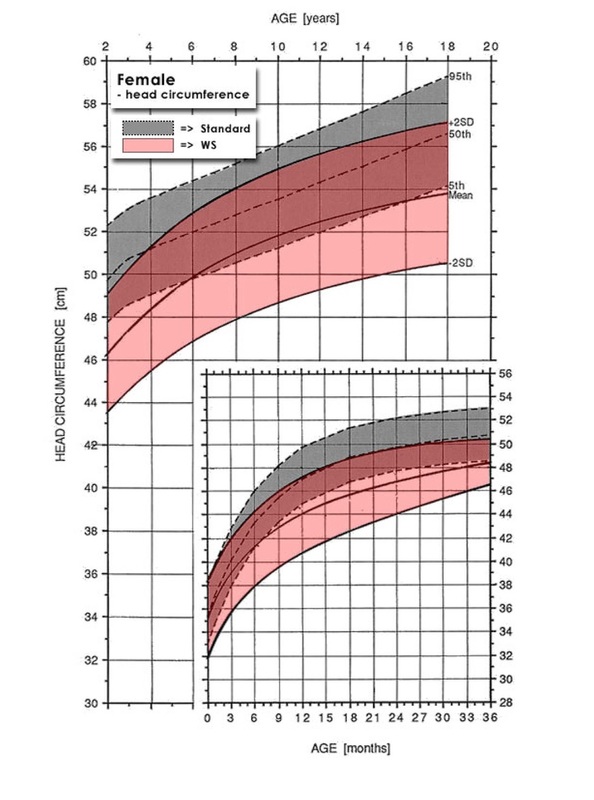
WS growth chart for females.
|
|
A cardiologist should evaluate individuals with Williams Syndrome yearly. This examination should include measurement of blood pressure in all four limbs and an echocardiogram of the heart.
An echocardiogram is a special form of ultrasound that looks at the structure of the heart. Doppler flow studies, which look at how the blood flows into and out of the heart, should also be done. Individuals with supravalvar stenosis may require surgery to fix this condition. The high blood pressure caused by this condition may be treated with medication. Examinations should take place yearly as some of these conditions are progressive and may worsen over time. |
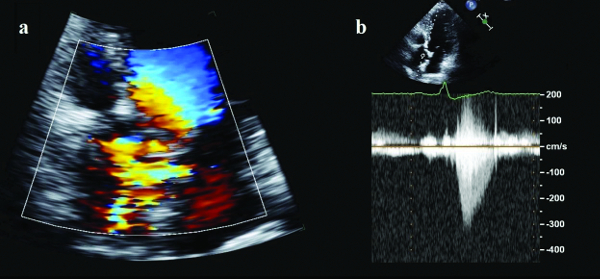
Transthoracic Echocardiography showing: a) supravalvular aliasing and acceleration of the flow at the Color-Doppler and b) a mild supravalvular aortic stenosis with the Continuous-wave Doppler.
|
Individuals with Williams Syndrome should also have a complete neurological examination.
|

|
|
In addition, the blood calcium levels of individuals with Williams Syndrome should be monitored every two years. High levels of calcium can cause irritability, vomiting, constipations and muscle cramps. An individual found to have a high level of calcium should consult a nutritionist to make sure that their intake of calcium is not higher than 100% of the recommended daily allowance (RDA). Vitamin D can increase calcium levels in individuals with Williams Syndrome and therefore should not take multivitamins containing vitamin D. If calcium levels remain high after limiting vitamin D and decreasing dietary intake of calcium, an individual with hypercalcemia should see a nephrologist for further management and to monitor kidney function.
Professionals should also take extra care of children with Williams Syndrome when in the sun – extra sunscreen must be applied to the children to minimize their intake of vitamin D. |
|
|
As children with Williams Syndrome present with a number of dental abnormalities, most children will need to have these abnormalities corrected to assist with breathing, feeding or speech. Many of these dental changes require and are able to be corrected by a dentist or orthodontist.
|
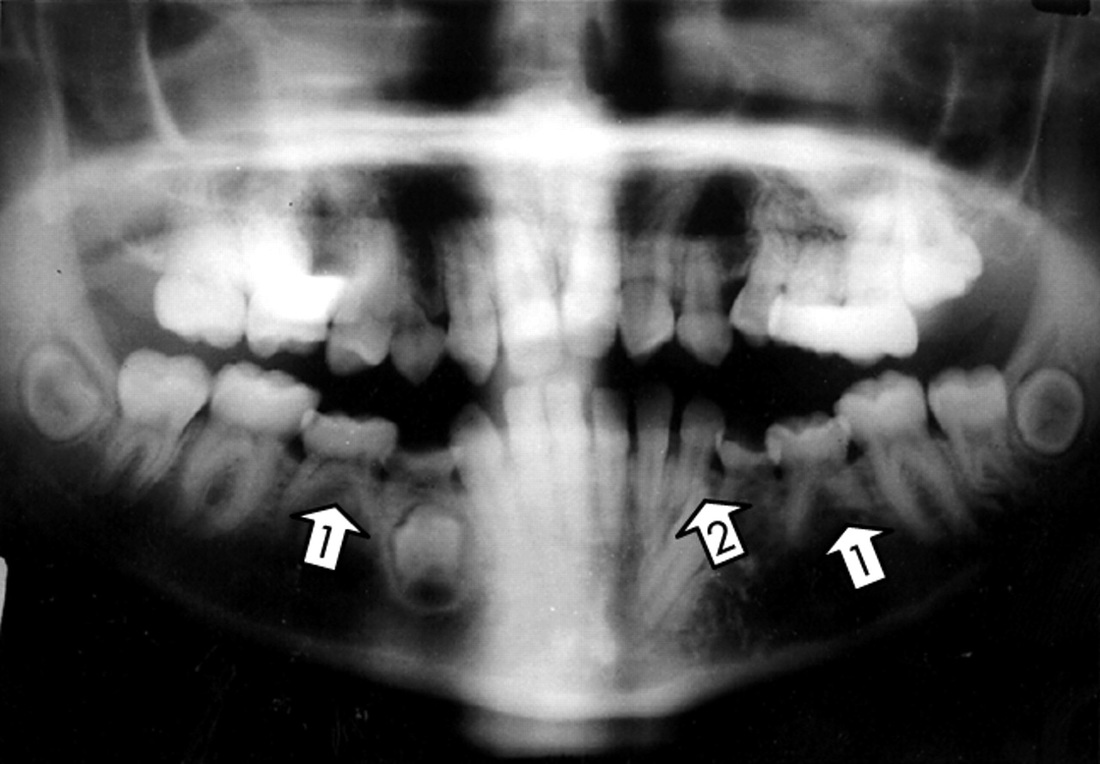
Orthopantomogram tomographic radiography of a typical Williams syndrome patient showing aplasia and tooth resorption anomaly, with fan shaped positioning of the front teeth.
|
|
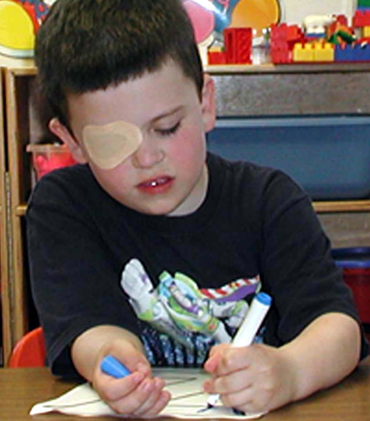
Strabismus (crossed eyes) can be treated by patching or by surgery.

|
Ear infections can be treated with antibiotics and surgical placement of ear tubes.
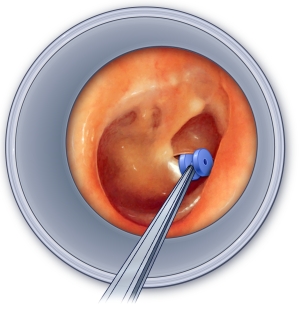
Tympanostomy tube (T-tube) -- the small blue piece -- being placed in a retracted ear drum.
|
|
The developmental differences of individuals with Williams Syndrome should be treated with early intervention and special education classes. Specific learning strategies that capitalise on the strengths of individuals with Williams Syndrome should be used. Physical, occupational, and speech therapy should be provided. Behavioral counselling and medication may help with behavioral problems such as hyperactivity and anxiety.
Children with Williams Syndrome often present with low muscle tone and as they get older, joint stiffness (contractures) may develop. Physical therapy is very helpful in improving muscle tone, strength and joint range of motion. Children with William Syndrome sometimes have sensitive hearing which often improves with age however may require the care of an ENT specialist if further complications arise. As they grow, children with Williams Syndrome also struggle with things like spatial relationships, numbers, and abstract reasoning, which can make daily tasks a challenge. Speech Pathologists are able to help individuals who have difficulty with spatial and semantic relationships. Occupational Therapists and Psychologists are also involved in assisting with numeracy and abstract reasoning difficulties. Speech Pathologists are most often involved in the social training of children with Williams Syndrome. Although they are extremely sociable and experience the need to connect with others; they are often overly social and have difficulty process nuanced social cues making it difficult to form lasting relationships. A speech pathologist will individually tailor a social training program based on the child’s social level, to assist with everyday common social interactions. |

|

|

|

SPEECH THERAPY:
The overall goal of PROMPT treatment is greater speech intelligibility and improved overall communication skills.
|
Currently research is being done with brain mapping and in the future it may be possible to tailor-make interventions, either preventative or palliative by understanding how a tiny change in the structure of a protein can lead to both anatomical and behavioral differences (Brynie, 2012).

Salk.edu,. 'Williams Syndrome - Salk Institute'. N.p., 2015. Web. 18 July 2015.
|
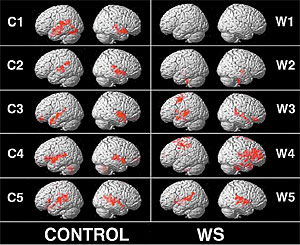
Levitin, D. J., Menon, V., Schmitt, J. E., Eliez, S., White, C. D., Glover, G. H., ... & Reiss, A. L. (2003). Neural correlates of auditory perception in Williams syndrome: an fMRI study. Neuroimage, 18(1), 74-82.
|

|
Prevention |
|
Williams Syndrome (WS) is not a disease and cannot be caught or cured.
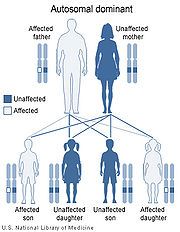
There is no known prevention for Williams syndrome. Although a genetic link is present, most babies diagnosed with Williams Syndrome will have no known family connection. According to the National Institute of Neurological Disorders and Stroke, random genetic mutations, not heredity, usually cause the condition. Even parents who have a child with WS have the exact same odds when they have another child. However, if a person with Williams Syndrome goes on to have children, his or her child has about a 50 percent chance of having the syndrome. Only one parent needs to have WS for the possibility of the children to inherit the faulty chromosome. (Scherer, 2005) Individuals with a family history of the condition can choose to undergo genetic counseling before conceiving a child. |
|

|
Possible Misdiagnosis
|
William's Syndrome "should be distinguished from other syndromes that include developmental delay, short stature, distinctive facies, and congenital heart disease."
Syndromes that should be distinguished from William's Syndrome include:
Marshall-Smith Syndrome similarities include mature bones, problems with growth and nutrition, ear infections, small chin and upturned nose, mental and physical delays.
Syndromes that should be distinguished from William's Syndrome include:
- Noonan Syndrome
- Deletion 22q11 (DiGeorge syndrome)
- Smith-Magenis syndrome
- Kabuki syndrome
- Fetal Alcohol Syndrome (FAS)
Marshall-Smith Syndrome similarities include mature bones, problems with growth and nutrition, ear infections, small chin and upturned nose, mental and physical delays.
- Supravalvar aortic stenosis, an autosomal dominant disorder characterized by elastin arteriopathy, is caused by mutation or intragenic deletions of ELN resulting in loss of function.
- Autosomal dominant cutis laxa, a primarily cutaneous condition, is the result of frameshift mutations at ELN that cause a dominant-negative effect on elastic fiber structure.

|
References |
Bayés M, Megano LF, Rivera N, Flores R, Pérez Jurado LA. (2003). Mutational mechanisms of Williams-Beuren syndrome deletions. Am J Hum Genet. 73, 131–151.
Brynie, F. (2012). Brain Architecture and Williams Syndrome. Psychology Today. Retrieved 27 July 2015, from https://www.psychologytoday.com/blog/brain-sense/201203/brain-architecture-and-williams-syndrome
Cornish, K., & Wilding, J. (2010). Attention, genes, and developmental disorders. Oxford: Oxford University Press.
Cuscó, I., Corominas, R., Bayés, M., Flores, R., Rivera-Brugués, N., Campuzano, V., & Pérez-Jurado, L. A. (2008). Copy number variation at the 7q11. 23 segmental duplications is a susceptibility factor for the Williams-Beuren syndrome deletion. Genome research, 18(5), 683-694.
Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, et al. (1993). Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 5, 11–16.
Frangiskakis JM, Ewart AK, Morris CA, Mervis CB, Bertrand J, et al. (1996). LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell. 86, 59–69.
Genetic Science Learning Center. What are Genetic Disorders?. Learn.Genetics. Retrieved July 27, 2015, from http://learn.genetics.utah.edu/content/disorders/chromosomal/williams/
Hirota H, Matsuoka R, Chen XN, Salandanan LS, Lincoln A, et al. (2003). Williams syndrome deficits in visual spatial processing linked to GTF2IRD1 and GTF2I on chromosome 7q11.23. Genet Med. 5, 311–321.
Hobart HH, Morris CA, Mervis CB, Pani AM, Kistler DJ, et al. (2010). Inversion of the Williams syndrome region is a common polymorphism found more frequently in parents of children with Williams syndrome. Am J Med Genet C Semin Med Genet. 154C, 220–228.
Jabbi, M., Kippenhan, J. S., Kohn, P., Marenco, S., Mervis, C. B., Morris, C. A., ... & Berman, K. F. (2012). The Williams syndrome chromosome 7q11. 23 hemideletion confers hypersocial, anxious personality coupled with altered insula structure and function. Proceedings of the National Academy of Sciences, 109(14), E860-E866.
Lowery MC, Morris CA, Ewart A, et al. (1995). Strong correlation of elastin deletions, detected by FISH, with Williams syndrome: evaluation of 235 patients. Am J Hum Genet. 57(1), 49-53.
Metcalfe K, Rucka AK, Smoot L, Hofstadler G, Tuzler G, et al. (2000). Elastin: mutational spectrum in supravalvular aortic stenosis. Eur J Hum Genet. 8, 955–963.
Morris CA. (2010a). The behavioral phenotype of Williams syndrome: a recognizable pattern of neurodevelopment. Am J Med Genet C Semin Med Genet. 154C, 427–431.
Morris CA. (2010b). Introduction: Williams syndrome. Am J Med Genet C Semin Med Genet. 154C, 203–208.
Morris CB, Mervis CA. (2000). Wiliams syndrome and related disorders. Annual Review of Genomics and Human Genetics. 1, 461–484.
Morris CA, Mervis CB, Hobart HH, Gregg RG, Bertrand J, et al. (2003). GTF2I hemizygosity implicated in mental retardation in Williams syndrome: genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am J Med Genet A. 123, 45–59.
Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, et al. (2001). A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 29,321–325.
Pober BR. (2010). Williams-Beuren syndrome. N Engl J Med. 362, 239–252.
Scherer SW, Gripp KW, Lucena J, Nicholson L, Bonnefont JP, et al. (2005). Observation of a parental inversion variant in a rare Williams-Beuren syndrome family with two affected children. Hum Genet. 117, 383–388.
Schubert C. (2009). The genomic basis of the Williams-Beuren syndrome. Cell Mol Life Sci. 66, 1178–1197.
Strømme P, Bjørnstad PG, Ramstad K. (2002). Prevalence estimation of Williams syndrome. J Child Neurol. 17, 269–271.
The University of Waikato. (2008). Chromosomes crossing over | Biotech Learning Hub. Biotechlearn.org.nz. Retrieved 27 July 2015, from http://biotechlearn.org.nz/focus_stories/evolved_enzymes/images/chromosomes_crossing_over
Van Hagen JM, van der Geest JN, van der Giessen RS, Lagers-van Haselen GC, Eussen HJ, et al. (2007). Contribution of CYLN2 and GTF2IRD1 to neurological and cognitive symptoms in Williams syndrome. Neurobiol Dis. 26,112–124.
Brynie, F. (2012). Brain Architecture and Williams Syndrome. Psychology Today. Retrieved 27 July 2015, from https://www.psychologytoday.com/blog/brain-sense/201203/brain-architecture-and-williams-syndrome
Cornish, K., & Wilding, J. (2010). Attention, genes, and developmental disorders. Oxford: Oxford University Press.
Cuscó, I., Corominas, R., Bayés, M., Flores, R., Rivera-Brugués, N., Campuzano, V., & Pérez-Jurado, L. A. (2008). Copy number variation at the 7q11. 23 segmental duplications is a susceptibility factor for the Williams-Beuren syndrome deletion. Genome research, 18(5), 683-694.
Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, et al. (1993). Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 5, 11–16.
Frangiskakis JM, Ewart AK, Morris CA, Mervis CB, Bertrand J, et al. (1996). LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell. 86, 59–69.
Genetic Science Learning Center. What are Genetic Disorders?. Learn.Genetics. Retrieved July 27, 2015, from http://learn.genetics.utah.edu/content/disorders/chromosomal/williams/
Hirota H, Matsuoka R, Chen XN, Salandanan LS, Lincoln A, et al. (2003). Williams syndrome deficits in visual spatial processing linked to GTF2IRD1 and GTF2I on chromosome 7q11.23. Genet Med. 5, 311–321.
Hobart HH, Morris CA, Mervis CB, Pani AM, Kistler DJ, et al. (2010). Inversion of the Williams syndrome region is a common polymorphism found more frequently in parents of children with Williams syndrome. Am J Med Genet C Semin Med Genet. 154C, 220–228.
Jabbi, M., Kippenhan, J. S., Kohn, P., Marenco, S., Mervis, C. B., Morris, C. A., ... & Berman, K. F. (2012). The Williams syndrome chromosome 7q11. 23 hemideletion confers hypersocial, anxious personality coupled with altered insula structure and function. Proceedings of the National Academy of Sciences, 109(14), E860-E866.
Lowery MC, Morris CA, Ewart A, et al. (1995). Strong correlation of elastin deletions, detected by FISH, with Williams syndrome: evaluation of 235 patients. Am J Hum Genet. 57(1), 49-53.
Metcalfe K, Rucka AK, Smoot L, Hofstadler G, Tuzler G, et al. (2000). Elastin: mutational spectrum in supravalvular aortic stenosis. Eur J Hum Genet. 8, 955–963.
Morris CA. (2010a). The behavioral phenotype of Williams syndrome: a recognizable pattern of neurodevelopment. Am J Med Genet C Semin Med Genet. 154C, 427–431.
Morris CA. (2010b). Introduction: Williams syndrome. Am J Med Genet C Semin Med Genet. 154C, 203–208.
Morris CB, Mervis CA. (2000). Wiliams syndrome and related disorders. Annual Review of Genomics and Human Genetics. 1, 461–484.
Morris CA, Mervis CB, Hobart HH, Gregg RG, Bertrand J, et al. (2003). GTF2I hemizygosity implicated in mental retardation in Williams syndrome: genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am J Med Genet A. 123, 45–59.
Osborne LR, Li M, Pober B, Chitayat D, Bodurtha J, et al. (2001). A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 29,321–325.
Pober BR. (2010). Williams-Beuren syndrome. N Engl J Med. 362, 239–252.
Scherer SW, Gripp KW, Lucena J, Nicholson L, Bonnefont JP, et al. (2005). Observation of a parental inversion variant in a rare Williams-Beuren syndrome family with two affected children. Hum Genet. 117, 383–388.
Schubert C. (2009). The genomic basis of the Williams-Beuren syndrome. Cell Mol Life Sci. 66, 1178–1197.
Strømme P, Bjørnstad PG, Ramstad K. (2002). Prevalence estimation of Williams syndrome. J Child Neurol. 17, 269–271.
The University of Waikato. (2008). Chromosomes crossing over | Biotech Learning Hub. Biotechlearn.org.nz. Retrieved 27 July 2015, from http://biotechlearn.org.nz/focus_stories/evolved_enzymes/images/chromosomes_crossing_over
Van Hagen JM, van der Geest JN, van der Giessen RS, Lagers-van Haselen GC, Eussen HJ, et al. (2007). Contribution of CYLN2 and GTF2IRD1 to neurological and cognitive symptoms in Williams syndrome. Neurobiol Dis. 26,112–124.
Home > Terminology > Characteristics > Causes, Diagnosis, Treatment > Prevalence & Early Warning Signs
Tips for Teachers > Classroom Management > Writing & Reading > Mathematics > Music
Tips for Teachers > Classroom Management > Writing & Reading > Mathematics > Music
Last Modified: 30th July 2015
